神奈川県行政書士会薬事相談員の横山です。
医療機器の輸出について、欧州向けの規制の概要をお知らせします。
海外→日本への輸入においては、日本の法令に適合する必要がありましたね。
また、日本で新規に医療機器を開発して、国内で発売する際にも同様です。
日本で製造した機器を欧州に輸出する場合も、様々な規制があります。
ちなみに、アメリカFDAの手続はこちらです。


CEマーキングとは?
欧州のCEマーキングとは、欧州連合(EU)内で売買される製品について「EU内で売ってよろしい」という販売許可のことで、一箇所で適合すれば加盟各国で有効です。
玩具やレジャー用船舶、花火にもCEマーキングの取得が義務づけられています。
CEとは「Conformite Europeene」を意味し、マークではなくマーキングの方が正式で、考え方としては事業者自身が適合させるもので、日本でいう製品の認証とは異なります。
概要
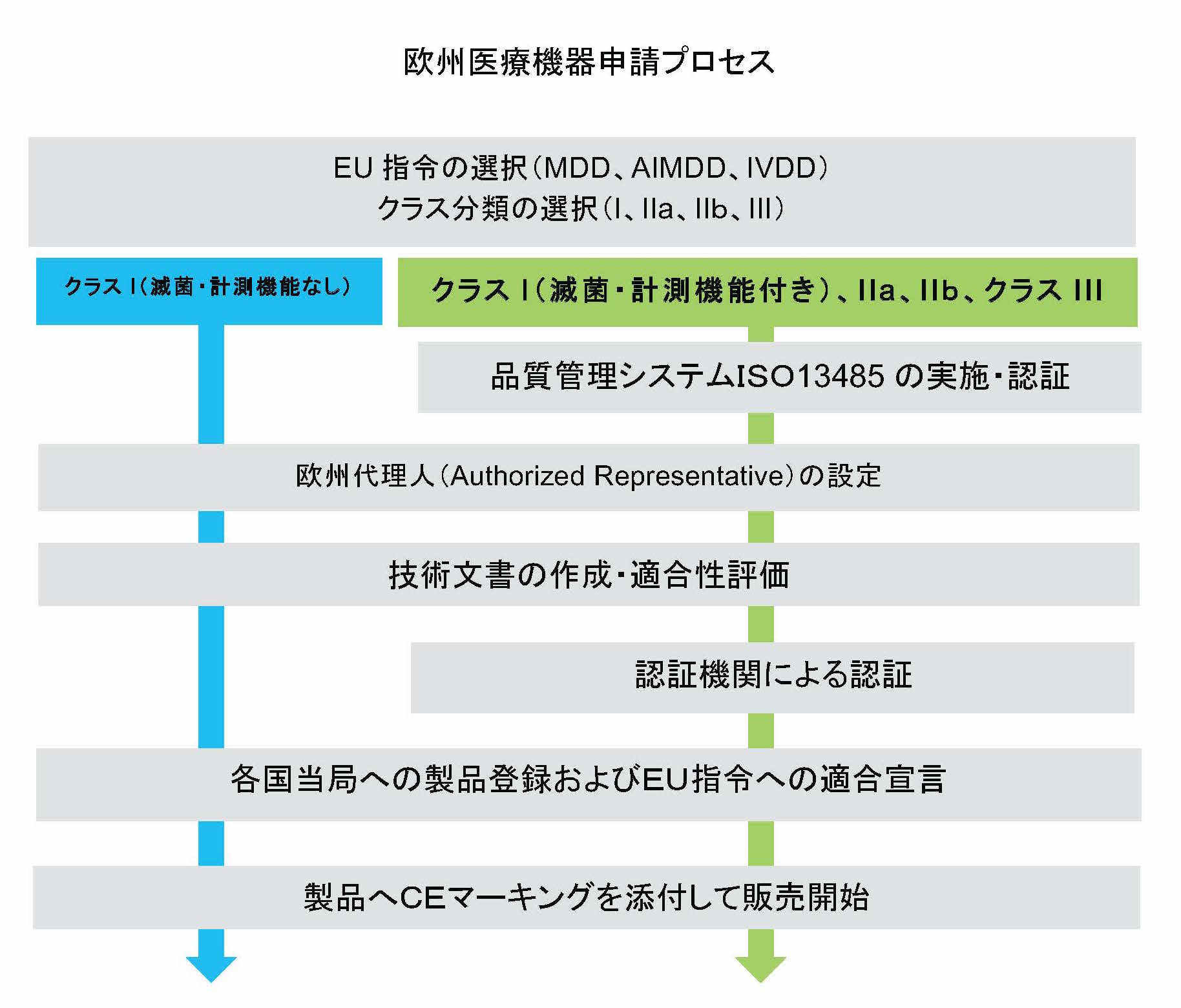
医療機器の輸入販売における手続は、欧州のどの国でも同様ですが
①CEマーキングの取得
②所轄州官庁への医療機器流通開始届
を経て販売できるようになる、というのが大枠です。
流れとしては、EU指令という欧州内共通の規制に適合することで、CEマーキングを取得します。
その後、販売開始する国(地域)で届出をしますが、欧州内での責任者の設置や製造物責任に関する規制などがあり、ほぼどこでも同様のようです。
医療機器の分類
欧州では、医療機器はリスクの低い順にクラスI、IIa、IIb、IIIの4つに分類されます。
人体に対する使用期間、体内への侵襲性、電気エネルギーの使用、中枢循環器および神経系への適用を考慮して、リスク分類が決まります。
CEマーキングは基本的に自己適用ですが、リスクの高いクラスIIa以上の機器にはEUの指定機関による適合性評価が求められています。
この機関が関与することで、機器の安全性や有効性が監視される仕組みになっています。

技術文書
自己評価で良いクラスIも含め、全ての医療機器において技術文書(Technical Documents)を作成し、保管する義務があります。
技術文書とは、EU指令における要求事項を遵守していることを示す文書で、具体的には製品の図面やリスク分析結果、取扱説明書などが含まれます。
EU指令には、日本と同様に医療機器の「基本要件」もあります。
適合宣言書
EUの指令に適合していることを、自ら宣言する文書が適合宣言書になります。
大まかな手続は以上になりますが、欧州の医療機器の規制の考え方は、政府が直接管理する日本の規制とは基本的に異なります。
しかし、リスクの高い製品には政府指定の認証機関の利用を義務づけるなど間接的に関わることで、事業者の自主性を保持しつつ監視もできる仕組みになっている印象です。