神奈川県行政書士会薬事相談員の横山です。
前回の欧州に引き続き、アメリカへ医療機器を輸出する際の手続についてまとめました。
欧州編はこちらです。

概要
米国の医療機器規制は、政府主導で行われる点で、日本と考え方が似ています。
まずクラス分類を決定し、リスクの低い機器は既発品と同等性の評価をし(510Kと呼ばれる市販前届出)、リスクの高い機器には事前承認審査(PMA=Pre market Approval)があります。
製造者や輸入業者の施設登録や機器登録、QSR(Quality System Regulation)への適合を経て、市販できるようになります。
クラス分類
クラス分類は、機器に欠陥や不具合があった場合のリスクに応じて、I、II、IIIとなります。
FDAの医療機器データベースにアクセスして確認しますが、どのクラスに該当するか正確にわからない場合は、FDAに事前相談をすることもできます。
510KとPMAとは?
クラス分類が完了したら、既存製品との同等性の確認をします。
この手続を510Kといい、自社既存製品の一部変更につかう510K Specialや、FDAで認証基準が存在する製品のための510K Abbreviatedといったものもあり、こちらに該当する場合は審査期間が短くなります。
既存製品と同一性が認められないと、PMA(臨床試験を含む事前承認)が求められます。
ただ機器によっては、同一性があってもPMAに該当したり同一性がなくても510Kで済む場合もあり、先のクラス分類によって求められる要求事項が変わってきます。
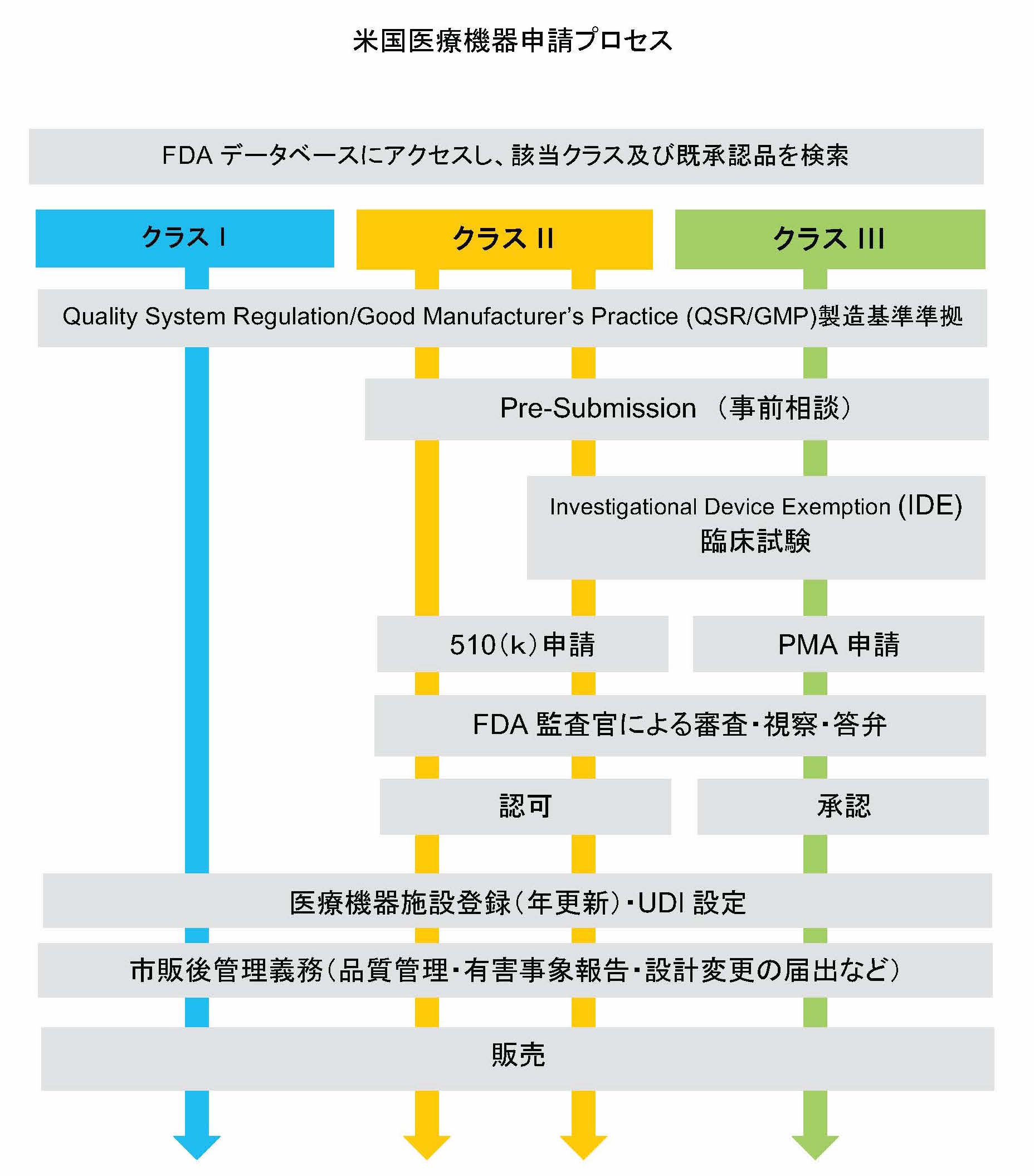

QSRの適合
QSR(Quality Sysytem Regulation)は、日本のQMSとほぼ同じでISO13485と類似しています。
ただ相違点もあるため、同じ機器を米国、日本、欧州で展開する場合には、QSR、QMS、ISO13485全てにおける配慮が必要になります。
今回、米国の医療機器規制について調査し、日本と大きく異なる点は、審査期間だと思いました。
クラスIIIの公表されている審査期間が、日本では1〜2年であるのに対し、米国では10〜12ヶ月で済むのですね。
医療機器の開発製造は成長が望める分野として政府が力を入れていますので、品質や安全確保に努めつつ、迅速な審査をしてくれることと思います。



この記事を読んだ人はこちらの記事も読んでいます